






The FDA hopes to review and act on 60 percent of prior approval supplement submissions in fiscal year 2015, 75 percent of PAS submissions in fiscal year 2016 and 90 percent of PAS submissions in fiscal year 2017.
The Generic Drug User Fee Amendments of 2012 (GDUFA) were signed into law on July 9, 2012, in an effort "to speed access to safe and effective generic drugs to the public and reduce costs to industry." In July 2014, the U.S. Food and Drug Administration (FDA) issued two draft Guidances for Industry: one relating to Prior Approval Supplements Under GDUFA and one relating to Amendments and Easily Correctable Deficiencies Under GDUFA.
Prior Approval Supplements Under GDUFA
Prior approval supplements (PASs) and amendments to PASs for abbreviated new drug applications (ANDAs) are submitted under section 505(j) of the Federal Food, Drug, and Cosmetic Act (the FD&C Act) (21 U.S.C. 355(j)). The FDA issued the following performance metric goals for PASs submissions:
- If an inspection is not needed, the goal is six months from the date of submission.
- If an inspection is needed, the goal is 10 months from the date of submission.
The FDA hopes to review and act on 60 percent of PAS submissions in fiscal year 2015, 75 percent of PAS submissions in fiscal year 2016 and 90 percent of PAS submissions in fiscal year 2017. However, these performance metric goals apply only to ANDA holders that submit a PAS on or after October 1, 2014. They do not apply to an amendment to a PAS if the PAS was submitted prior to that date. These performance metric goals also do not apply to new drug applications or biologics license applications or to supplements thereto. If an amendment is made to a PAS, the GDUFA goal date may be revised by the FDA.
The submission date is defined as the date that the PAS arrives in the appropriate electronic portal of the FDA. The FDA will then count this submission date as the first day of the review period and will calculate the goal date in months. If FDA refuses to receive a PAS, the GDUFA review clock will stop. The applicant can then submit a corrected or new supplement, but a new GDUFA fee will be required and a new review clock will start, resulting in a new goal date for that PAS.
The following events can result in the FDA's refusal to receive a PAS:
- failure to pay the application fee within 20 calendar days of submission;
- reference to a drug master file (DMF) that is not on the public available for reference list;
- reference to a facility on the facility arrears list;
- the applicant is the owner or is affiliated with the owner of a facility on the facility arrears list; or
- the applicant is on or affiliated with an entity on the backlog arrears list.
If a PAS is substantially complete except for failure to pay the PAS user fee, the PAS will be deemed received when the fee is paid. Similarly, if a PAS is substantially complete but references a facility on the arrears list, the PAS will be deemed received once the facility is removed from that list.
Determining whether an inspection is required for a PAS is within the discretion of the FDA and depends, in part, on the nature of the supplement. If the PAS involves a facility not approved in the original ANDA or if it involves a fundamental change to the manufacturing process or technology, an inspection may be required. FDA, however, will rely on previous inspections of a finished product site if it occurred within two years of the current Good Manufacturing Practice (cGMP) evaluation for a pending application, three years for an active pharmaceutical ingredient (API) site or control testing laboratory and four years for a packaging-only site. Some exceptions may apply to this general practice, depending on the nature of the drug being processed or the complexity of the processing operations.
The FDA considers multiple supplements submitted to multiple ANDAs by a single applicant for the same chemistry, manufacturing and controls change to each ANDA to be a "grouped supplement." However, each supplement in this group is considered an individual submission and requires its own GDUFA fee for each ANDA in the group. That being said, the submissions will generally have the same GDUFA goal date.
If a supplement is filed as a Changes Being Effected (CBE) but the FDA finds that such submission should have been submitted as a PAS, the FDA will notify the applicant and then administratively close and withdraw the CBE. The applicant may then resubmit the supplement as a PAS, along with the required GDUFA fee. The GDUFA review clock will run from the date of submission of the PAS.
Amendments and Easily Correctible Deficiencies Under GDUFA
The FDA has issued the following performance metric goals for amendments submitted electronically to original ANDAs and PASs filed on or after October 1, 2014.
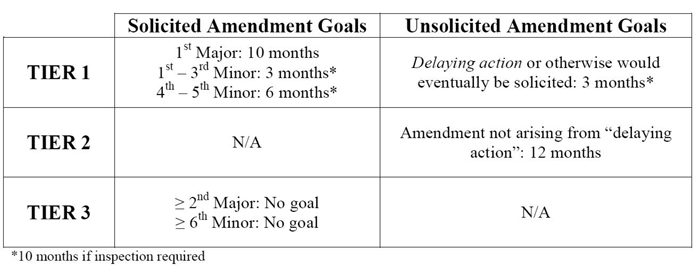
For original ANDAs or PASs submitted before October 1, 2014, these performance goals are inapplicable to subsequently filed amendments thereto. Where these goals apply, such amendments may add additional time to the original ANDA review, but in no case will they shorten the original goal date. The time periods above are calculated from the date of submission of the amendment.
"A solicited amendment is a submission made by an applicant in response to a complete response letter (CR) issued by FDA." These amendments are classified as either Tier 1 or Tier 3 and either as a major amendment, a minor amendment or an easily correctable deficiency (ECD). A major amendment "contain[s] a substantial amount of new data or new information not previously submitted to reviewed by FDA, requiring … a substantial expenditure of FDA resources." The first solicited major amendment is classified as Tier 1; any subsequent major amendment is classified as Tier 3. Appendix A of the Draft Guidance provides a nonexhaustive list of deficiencies that generally require major amendments. A minor amendment, on the other hand, "requires … fewer FDA resources than are necessary to review a major amendment but more than are necessary to review the information submitted in response to an ECD." For instance, a minor amendment may address missing information but not require any new studies to be performed. The first through fifth solicited minor amendments are classified as Tier 1; any subsequent minor amendment is classified as Tier 3. Appendix B of the Draft Guidance provides a nonexhaustive list of deficiencies that generally require minor amendments. Any Tier 3 amendment, whether major or minor, will cause the ANDA to lose its goal date. Finally, ECDs "require[] … a modest expenditure of FDA resources." They can be responded to quickly because the applicant should already have the necessary information. ECDs generally relate to requests for clarification, requests for postapproval commitments or final resolution of technical issues. ECDs do not extend the goal date. Appendix C of the Draft Guidance provides a nonexhaustive list of deficiencies that are generally ECDs.
"An unsolicited (gratuitous) amendment is submitted on the applicant’s own initiative and not in response to FDA's CR letter." These amendments are classified as either delaying or nondelaying. All delaying amendments are Tier 1 and all non-delaying amendments are Tier 2. A delaying amendment "address[es] actions by a third party that would cause delay or impede application review or approval timing and that were not a factor at the time of submission." Delaying amendments do not add to the count of major or minor amendments. A nondelaying amendment "contain[s] information that is not requested by FDA and is not the result of changes to the RLD or USP monograph, changes to the RLD labeling, a REMS and REMS modification, or generic approval requirements reflected in citizen petition responses issued by FDA."
By contrast, an administrative amendment is "routine in nature and do[es] not require scientific review." These types of amendments include "[r]equests for final approval with no scientific changes to the ANDA, patent amendments, and general correspondence submitted by applicants." These amendments do not affect goal dates.
An applicant may request reconsideration of an amendment classification. If an applicant seeks reconsideration of a CR amendment, a written request for a post-CR-letter meeting must be sent to the regulatory project manager within 10 business days of issuance of the CR letter. The applicant will then be required to submit meeting materials that adequately explain the nature of the request. The division will issue a decision within 10 business days. If the request is granted after an amendment has been submitted and a review is pending, the change in classification will not alter the goal date but will adjust the amendment count. If the amendment has not yet been submitted, it will be assigned the revised classification and corresponding goal date. If an applicant seeks reconsideration of a change in classification after submission of a CR amendment, the request should be made in writing within 10 business days from issuance of the goal letter and contain adequate information to explain the nature of the dispute. The division will make a decision on this request within 10 business days, and if rendered, such a change in classification will not alter the goal date but will adjust the amendment count. If an applicant disagrees with the decision on reconsideration, it may file a formal appeal of the decision.
The Draft Guidance provides a series of useful examples for how these amendment goals are applied. It also provides an amendment flow chart that graphically depicts the different types of amendments and their effect on the goal date.
For Further Information
If you have any questions about this Alert, please contact Frederick (Rick) R. Ball, Carolyn A. Alenci, any member of the Pharmaceutical, Medical Device, Pharmacy & Food industry group or the attorney in the firm with whom you are regularly in contact.
Disclaimer: This Alert has been prepared and published for informational purposes only and is not offered, nor should be construed, as legal advice. For more information, please see the firm's full disclaimer.